
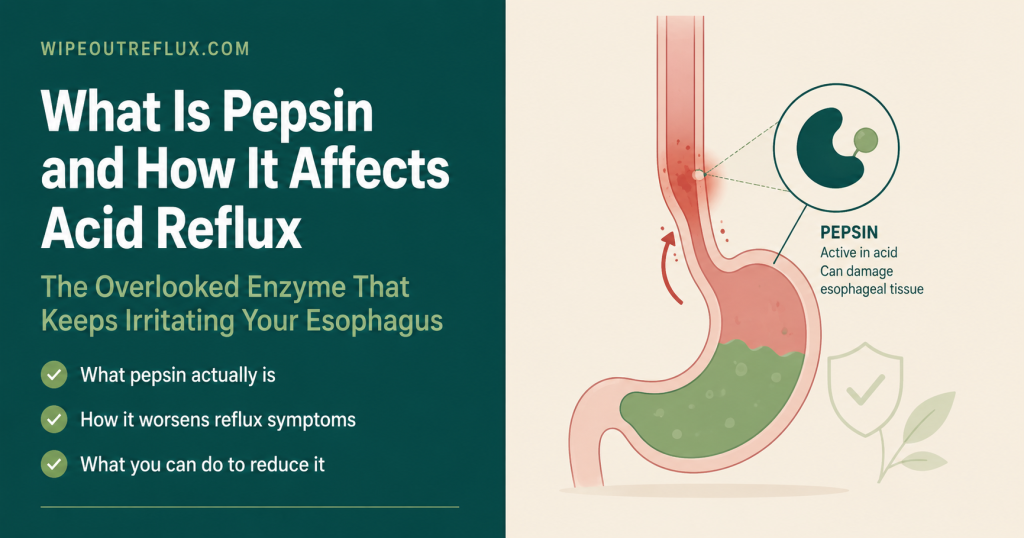
If you have acid reflux or LPR, pepsin is the most important thing you’ve probably never heard enough about. Most people have a basic understanding of stomach acid and heartburn. Fewer understand that the enzyme being released alongside that acid — pepsin — is, in many cases, the primary agent causing tissue damage, and that it operates in ways that acid suppression alone simply cannot address.
Pepsin is a digestive enzyme produced exclusively in the stomach. Its normal job is to break down dietary protein. The problem begins when it travels out of the stomach via reflux: into the oesophagus in GERD, and into the throat, larynx, and beyond in LPR (silent reflux). Once it reaches these tissues, pepsin behaves in ways that are both more persistent and more damaging than acid alone — and that are not resolved by PPIs, because PPIs reduce acid without touching the enzyme.
Understanding pepsin — how it’s made, when it’s active, what it does to tissue, and how it can be deactivated — is the key to understanding why reflux management, particularly for LPR, requires more than an acid-suppressing medication. It also explains why what you eat and drink after a reflux event matters just as much as preventing the reflux in the first place.
Key Takeaways
- Pepsin is a proteolytic (protein-digesting) enzyme produced exclusively in the stomach. It begins as an inactive precursor called pepsinogen, which is released by gastric chief cells and converted to active pepsin by the low pH of stomach acid.
- Pepsin is most active at pH 1.5–3.0 — the typical pH of the stomach. Its activity diminishes as pH rises, but it remains stable (dormant, not destroyed) at neutral pH 7.4, and can be reactivated by any subsequent drop in pH — including from dietary acid.
- Complete irreversible inactivation of pepsin only occurs above pH 8 — which is why alkaline water at pH 8.8 permanently destroys the enzyme, while ordinary neutral water (pH 6.7–7.4) leaves it intact.
- Laryngeal tissue is essentially resistant to acid damage at pH 4.0, but is significantly damaged when pepsin is present at the same pH — establishing pepsin, not acid alone, as the primary damaging agent in LPR.
- Pepsin doesn’t just sit on the surface of laryngeal tissue after a reflux event — it is actively taken up into cells by receptor-mediated endocytosis, where it causes intracellular damage even at neutral pH, from inside the cells that have absorbed it.
- Pepsin depletes the larynx’s own protective proteins (carbonic anhydrase III, Sep70) and degrades E-cadherin — the protein that holds epithelial cells together — compromising the mucosal barrier and driving chronic inflammation.
- PPIs reduce acid production but do not inactivate or remove pepsin. Patients on PPIs may have less acidic reflux but the pepsin in that reflux remains active — explaining why PPI therapy often fails to resolve LPR throat symptoms.
- Pepsin can be detected in saliva, tears, middle ear fluid, and other sites using the Peptest lateral flow device — confirming reflux has reached those tissues. Pepsin detected outside the stomach is a biomarker that only reflux can explain.
- Alginates physically block pepsin from reaching the oesophagus and throat by forming a raft above stomach contents, and directly inhibit pepsin enzymatic activity — providing protection that acid suppression alone cannot.
What Is Pepsin? The Basics
Pepsin is an aspartic protease — a class of enzymes that break down proteins by cleaving the peptide bonds between amino acids. It is one of the most powerful protein-digesting enzymes in the human digestive system, and in its intended context — the stomach — it is essential for the early stages of protein digestion.
The enzyme was first described in 1836 by Theodor Schwann, who identified it as the substance in gastric juice capable of digesting proteins. Its name comes from the Greek word for digestion: pepsis. It was among the first enzymes ever to be crystallised (by John H. Northrop in 1928), making it one of the most historically studied enzymes in biochemistry.
Structurally, pepsin is a single polypeptide chain that acts as a highly efficient molecular scissors. Its active site contains two aspartic acid residues that work together to cleave the peptide bonds in proteins — particularly at sites adjacent to aromatic amino acids like tyrosine, phenylalanine, and tryptophan. This broad but specific cleavage pattern makes pepsin an excellent general-purpose protein-digesting enzyme for the stomach’s diverse dietary protein load.
What makes pepsin relevant to reflux — and specifically to LPR — is not its normal function in the stomach. It’s what happens when it travels to tissues it was never designed to reach.
How Pepsin Is Made: The Pepsinogen to Pepsin Conversion
Pepsin doesn’t exist as an active enzyme in the stomach wall. If it did, it would digest the stomach’s own cells. Instead, it’s stored and secreted in an inactive form called pepsinogen — a precursor molecule that is chemically locked and cannot digest proteins until it encounters the right conditions.
Pepsinogen is synthesised and stored in granules within the chief cells (also called peptic cells or zymogenic cells), which are located in the deeper layers of the gastric glands in the stomach’s fundus and body. Chief cells are stimulated to release pepsinogen by three main signals: acetylcholine (the neurotransmitter released by the vagus nerve during the anticipation and early stages of eating), gastrin (a hormone released by G-cells in the stomach antrum in response to protein in the stomach), and the low pH of the stomach itself.
When pepsinogen is secreted into the gastric lumen, it encounters the highly acidic environment created by the parietal cells (which pump hydrogen ions to produce hydrochloric acid, lowering pH to around 1.5–3.5). This acidity triggers a conformational change in the pepsinogen molecule, cleaving off an inhibitory portion and revealing the active site. The result is active pepsin — and the process is autocatalytic: once some pepsin is active, it can also convert additional pepsinogen molecules to pepsin, amplifying the response.
This activation mechanism — requiring the very acid that comes from the stomach’s parietal cells — is important for reflux management. It explains why PPIs (proton pump inhibitors), which reduce acid production by blocking the hydrogen pumps in parietal cells, also indirectly reduce pepsin activity in the stomach: less acid means less pepsinogen activation. But it does not explain how pepsin is addressed once it has already been activated and has already left the stomach. That’s a different problem entirely.
Pepsin’s Normal Role: Protein Digestion in the Stomach
In its intended context, pepsin performs a specific and essential digestive function. When you eat a protein-containing meal, pepsin begins the process of unfolding and fragmenting dietary proteins into smaller units called polypeptides — fragments that can then be further digested by other enzymes (trypsin, chymotrypsin, elastase) in the small intestine, before finally being broken into individual amino acids for absorption.
Pepsin doesn’t complete protein digestion — it starts it. Its contribution is particularly important for proteins that have a tightly folded (globular) structure, which are harder for enzymes to access. Pepsin’s initial cleavage opens up these structures, making the fragments accessible for the next stage of digestion.
The stomach is uniquely designed to contain pepsin safely. Gastric mucus forms a protective barrier on the stomach’s inner surface, and the mucus layer is continuously renewed to protect the epithelial cells from both acid and pepsin. The stomach’s cells also renew themselves at remarkable speed — the gastric pit cells are replaced every 2–4 days — in part as a protective mechanism against the very enzymes they produce.
The oesophagus has some acid-protective mechanisms but fewer than the stomach. The throat and larynx have essentially none at all. This asymmetry is the biological foundation of the reflux damage problem.
What Happens When Pepsin Leaves the Stomach
When reflux occurs — whether the full-oesophageal variety of GERD or the upper-throat variety of LPR — active pepsin leaves the stomach along with acid and other gastric contents. Once outside the stomach, pepsin enters tissues for which it has no business being there, with no protective mechanisms in place.
In GERD, pepsin enters the oesophagus. The oesophageal mucosa, while more robust than laryngeal tissue, is progressively damaged by pepsin’s proteolytic activity, particularly as pepsin works alongside acid to degrade the intercellular junctions that maintain the mucosal barrier. Pepsin degrades E-cadherin — a protein that bridges adjacent epithelial cells and maintains the tightness of the barrier between them. When E-cadherin is cleaved, the barrier becomes more permeable, and acid, pepsin, and other irritants can penetrate more deeply into the tissue, driving the chronic inflammation that characterises GERD-related oesophagitis and, over time, contributing to Barrett’s oesophagus and the elevated cancer risk associated with long-standing reflux disease.
In LPR, pepsin travels further — past the upper oesophageal sphincter and into the laryngopharynx: the throat, vocal cords, posterior commissure, subglottic area, and surrounding structures. The laryngeal mucosa has no equivalent of the gastric mucus barrier, no mucosal bicarbonate secretion system, and a much lower cell renewal rate than the gastric epithelium. It is profoundly ill-equipped to withstand the proteolytic activity of an enzyme designed to digest proteins at pH 2.
The relationship between pepsin, acid, and LPR is covered from the clinical perspective in the GERD vs LPR comparison — the two conditions differ substantially in their tissue targets, damage mechanisms, and therefore their treatment approaches.
The pH Activity Profile: Understanding Pepsin’s On-Off States
One of the most important things to understand about pepsin — and one of the things that most distinguishes LPR management from standard GERD management — is its pH-dependent behaviour. Pepsin doesn’t simply work at low pH and not at high pH. Its relationship with pH is more nuanced, and those nuances have direct implications for treatment.
The activity profile of human pepsin 3b (the dominant form) looks like this:
- pH 1.5–3.0: Peak activity. Pepsin is fully active and maximally efficient at digesting proteins. This is the normal stomach environment.
- pH 3.0–4.0: Activity decreasing but still significant — pepsin can still cause mucosal damage at these pH values when in contact with laryngeal tissue, as demonstrated by direct tissue exposure experiments.
- pH 4.0–6.5: Activity progressively diminishing. Pepsin is less enzymatically active but still capable of causing damage, particularly through receptor-mediated uptake into cells. At pH 6.5, pepsin activity is essentially abolished.
- pH 6.5–7.4: Pepsin is enzymatically inactive — but critically, it is not destroyed. It is dormant. It sits in the tissue, or in the tear film, or in ear fluid, or wherever else it has travelled, waiting. The mean pH of the laryngopharynx is 6.8 — the normal environment of the throat. In this environment, pepsin is inactive but entirely stable, and can remain so for hours.
- pH >8.0: Irreversible denaturation. Above pH 8, the pepsin molecule permanently unfolds and loses its structure. It cannot be reactivated. This is the basis for the alkaline water approach at pH ≥8.8, which permanently destroys pepsin on contact.
The dormancy at neutral pH is what makes LPR so persistent and so poorly addressed by acid suppression. The throat’s natural pH of 6.8 doesn’t inactivate pepsin — it preserves it in a stable, dormant state. And the next time anything acidic reaches the throat — another reflux event, a glass of orange juice, a bite of a citrus sweet — the pH drops below 6.5 again and pepsin reactivates.
Johnston et al. (2007) demonstrated this experimentally: after exposure of laryngeal tissue to pepsin at neutral pH (pH 7.4) for 24 hours, pepsin remained stable. When the pH was subsequently dropped to pH 3, 72% of peptic activity was recovered. The enzyme had been sitting dormant in the tissue, and the acidic challenge reactivated it. This is the reactivation mechanism that dietary management in LPR is designed to prevent — which is why eating anything below pH 5 matters even when no new reflux event has occurred.
The guide to neutralising pepsin in the throat covers the practical strategies for addressing pepsin at the tissue level.
Pepsin in the Oesophagus: GERD and Barrier Disruption
In GERD, the primary site of pepsin damage is the oesophageal epithelium. The mechanism is well characterised: pepsin, in conjunction with acid, degrades the structural proteins that maintain the integrity of the mucosal barrier.
The oesophageal mucosal barrier normally functions as a two-layer defence: an outer mucus layer that dilutes and buffers acid, and tight junctions between epithelial cells that prevent acid, pepsin, and other irritants from penetrating to deeper tissue. Pepsin attacks both layers. It enzymatically degrades the mucus proteins and — more damagingly — cleaves E-cadherin, the adhesion molecule that maintains tight junction integrity between epithelial cells.
When E-cadherin is cleaved by pepsin, the process is not simple protein degradation. Research has shown that E-cadherin undergoes a process called regulated intramembrane proteolysis (RIP): pepsin makes an initial cut at the cell surface, generating soluble E-cadherin fragments that then trigger downstream signalling, including the activation of matrix metalloproteinases (MMPs) — enzymes that further degrade the extracellular matrix. This is not just a structural problem; it’s the initiation of a molecular signalling cascade that promotes chronic inflammation, tissue remodelling, and over long periods, the cellular changes associated with Barrett’s oesophagus and oesophageal cancer risk.
Importantly, pepsin causes E-cadherin cleavage at weakly acidic pH (pH 4) — the kind of mildly acidic reflux that occurs in patients on PPIs who still have some acid reduction but not complete suppression. This is one of the key mechanisms by which reflux-associated oesophageal damage can progress even in patients who are taking PPIs and have reduced (but not eliminated) acid in their refluxate. The PPI reduces the acid; it doesn’t remove the pepsin, and the pepsin at pH 4 is still active enough to cause significant molecular damage.
Pepsin in the Throat: Why LPR Is Fundamentally a Pepsin Problem
If pepsin in the oesophagus is a significant problem, pepsin in the larynx is far worse — because the laryngeal mucosa is dramatically more vulnerable and has far fewer defences.
A landmark experimental study by Bulmer et al. (2010) directly quantified this vulnerability: porcine laryngeal tissue was exposed to either acid alone (pH 2 and pH 4) or to acid plus pepsin. At pH 4.0, acid alone caused essentially no significant tissue damage. But at pH 4.0 with pepsin present, significant tissue damage occurred across all tested laryngeal sites. The conclusion was clear and directly stated: laryngeal tissues are essentially resistant to damage at pH 4.0 alone, but are damaged when pepsin is present. In LPR, pH 4.0 or above refluxate would only be damaging if it contains pepsin.
This finding reframes the entire pathophysiology of LPR. The laryngeal damage associated with silent reflux — the hoarseness, the chronic throat clearing, the globus sensation, the posterior commissure hypertrophy, the vocal cord granulomas — is not primarily caused by acid. It’s caused by pepsin operating at the mildly acidic pH values that characterise proximal reflux events reaching the throat.
For a comprehensive overview of all the ways LPR differs from GERD and what symptoms it produces, the complete guide to LPR covers the clinical picture in full. The LPR symptoms guide covers the specific presentation — many of which are caused by pepsin damage to the precise tissues described here.
The Intracellular Discovery: Pepsin Gets Inside the Cells
Perhaps the most striking discovery in pepsin-LPR research came from Johnston et al. in 2006 and 2009: pepsin is not merely an extracellular irritant that sits on the surface of laryngeal tissue. It is actively taken up into laryngeal and hypopharyngeal cells by a process called receptor-mediated endocytosis.
This means that pepsin binds to specific receptors on the surface of laryngeal epithelial cells and is drawn inside the cell in membrane-bound vesicles called endosomes. Inside the cell, endosomes are naturally acidic — at around pH 5. And at pH 5, dormant pepsin that entered the cell at neutral pH is reactivated. It is now inside the cell, proteolytically active, and free to digest the cellular machinery around it.
The cellular consequences are specific and serious. Studies have documented:
- Mitochondrial swelling and dysfunction — disrupting the cell’s energy production
- Depletion of Sep70 and Sep53 — squamous epithelial stress proteins that protect cells under acid challenge. These are the larynx’s own stress-response protection, and pepsin specifically degrades them
- Disruption of carbonic anhydrase III (CAIII) — an enzyme that produces bicarbonate to neutralise acid locally; its depletion removes a key protective layer
- E-cadherin cleavage and downstream MMP upregulation — the same pathway described in the oesophagus, now operating from inside the cell
- Induction of proinflammatory cytokine gene expression — generating a sustained inflammatory response that persists beyond individual reflux events
The critical implication of receptor-mediated endocytosis is that pepsin can cause cellular damage at neutral pH — without needing acid to be present at the time of damage. Pepsin that entered the cell during a reflux event continues its damaging activity within the acidic endosomal environment long after the pH of the surrounding laryngeal tissue has returned to normal. This explains the chronic, progressive nature of LPR tissue injury and why symptoms persist even during periods when reflux seems to be well controlled.
Using Western blot analysis of laryngeal biopsies, Johnston et al. detected intracellular pepsin in 19 of 20 LPR patients confirmed by pH monitoring — and in only 1 of 20 healthy controls. This is about as close to a direct cellular fingerprint of LPR as the research literature currently provides.
Dietary Reactivation: Why What You Eat After a Reflux Event Matters
The dormancy of pepsin at neutral pH — and its reactivation by anything acidic — has a direct and important implication for dietary management in LPR: every acidic food or drink you consume after a reflux event has the potential to reactivate dormant pepsin in your throat, continuing the inflammatory cycle without a new reflux event.
This is the mechanistic basis for the LPR dietary recommendation to avoid foods and drinks below pH 5. At the mean laryngopharyngeal pH of 6.8, pepsin is dormant. Consuming something acidic — a coffee at pH 4.5–5, a fizzy drink at pH 2.7–3.9, citrus fruit at pH 3–4, a sour sweet at pH 1.8 — drops the laryngeal pH below 6.5 and reactivates the pepsin that’s already sitting in that tissue.
This dietary reactivation is why LPR symptoms often worsen after dietary triggers even in people who aren’t having new reflux events. The coffee doesn’t cause fresh reflux — it reactivates residual pepsin. The citrus fruit doesn’t produce new acid — it creates the pH environment that wakes up dormant enzymes in the laryngeal cells.
It also explains why the citric acid in foods and drinks is such a significant concern for LPR specifically — and why the LPR dietary framework is stricter about pH than the standard GERD framework. Reducing dietary acid isn’t just about not eating things that cause more reflux. It’s about not reactivating the enzyme that’s already sitting in your throat.
Why PPIs Don’t Address Pepsin
Proton pump inhibitors are the most prescribed class of drugs for acid reflux in the world, and they are highly effective at what they do: reducing the output of hydrogen ions by parietal cells, thereby reducing gastric acid production and raising intragastric pH. For erosive oesophagitis and ulcer healing, this is genuinely effective treatment. For GERD broadly, symptom relief is significant in many patients.
But PPIs have a specific and important limitation that explains why they so consistently fail patients with LPR: they do not address pepsin.
Pepsin that has already been activated and secreted into the stomach is not inactivated by PPIs. The PPI raises the pH of the stomach (and therefore makes future pepsin activation less efficient), but it cannot remove or destroy pepsin molecules already in circulation. When reflux occurs in a PPI-treated patient, the refluxate typically has a higher pH than in an untreated patient — but it still contains pepsin, and that pepsin, at the mildly acidic pH values still present in the stomach of a PPI-treated patient, remains proteolytically active.
Research confirms this. Studies using combined pH-impedance monitoring have found that non-acidic and weakly acidic reflux events — the kind that occur on PPI therapy, where acid production is suppressed but not eliminated — still produce pepsin-containing refluxate that reaches the throat in LPR patients. Pepsin at pH 4 (common in PPI-treated patients) still causes E-cadherin cleavage and MMP induction. Pepsin taken up by cells at neutral pH still causes intracellular damage.
This is the fundamental reason why PPI therapy for LPR has been shown repeatedly to be no more effective than placebo for throat symptoms in randomised trials. The drug addresses the wrong component. Understanding this is the single most important step in moving from PPI dependence toward the multi-pronged approach that actually addresses LPR. The article on getting off PPIs and managing acid rebound covers this transition in practical terms.
How Pepsin Can Be Detected
The fact that pepsin is produced exclusively in the stomach gives it unique value as a biomarker: wherever pepsin is found outside the stomach, reflux is the explanation. There is no other source.
This principle has driven the development of salivary pepsin testing — the most accessible non-invasive method of detecting reflux. The Peptest device (RD Biomed, Hull, UK) uses lateral flow technology with highly specific monoclonal antibodies to detect and quantify pepsin in saliva samples. Because pepsin can only reach saliva via reflux events that have brought it up to the throat level, a positive Peptest result confirms that reflux has reached the proximal oesophagus or throat — independent of whether the reflux was acidic or non-acidic.
The diagnostic performance of Peptest is nuanced. In LPR specifically, fasting Peptest has shown 98% specificity — meaning when it’s positive in a fasting sample, reflux is almost certainly the explanation. Sensitivity is more modest (40–48%), meaning a negative result doesn’t rule out LPR. For practical diagnostic purposes, Peptest is most useful as a confirmatory tool — a positive result provides high confidence that reflux is occurring, while a negative result should be interpreted alongside clinical findings. The dedicated Peptest guide covers how to use and interpret the test in detail.
As covered in earlier articles on this site, pepsin has also been detected in tears, middle ear fluid, and nasal secretions — confirming reflux as the source and explaining the extraoesophageal manifestations of LPR including dry eye symptoms, ear problems, and sinus congestion.
How to Reduce Pepsin’s Damage: The Four-Layer Approach
Managing pepsin in LPR requires a different framework from standard acid suppression — one that addresses pepsin at each stage of its journey and activity cycle.
1. Reduce the frequency and extent of reflux events
Fewer reflux events means less pepsin reaching the throat in the first place. This is the foundation: dietary management, meal timing, positional strategies, and treating any underlying mechanical factors (LES weakness, hiatus hernia, delayed gastric emptying). The LPR diet framework is built around this principle.
2. Block pepsin at the gastro-oesophageal junction
Alginate-based products (Gaviscon Advance) form a physical raft above stomach contents, preventing pepsin from entering the oesophagus and throat with each reflux event. Alginates also directly bind and inhibit pepsin — reducing its enzymatic activity by up to 53.9% in in vitro studies — and sequester bile acids from the refluxate. This is pepsin management at the source, before it reaches vulnerable tissue.
3. Inactivate pepsin that has reached the throat
Water at pH ≥8.8 permanently and irreversibly inactivates pepsin — denaturing the enzyme so it can never be reactivated. Drinking alkaline water (or using alkaline water drops to raise pH) between meals and particularly after any dietary acid exposure provides an opportunity to destroy dormant pepsin in the throat before it can cause further damage. The alkaline water and LPR guide covers the evidence behind this strategy.
4. Avoid dietary reactivation
Keeping the throat environment above pH 5 prevents dormant pepsin from being reactivated. This means avoiding all foods and drinks below pH 4–5 during the active management phase — citrus, vinegar, acidic drinks, sour candy, and carbonated beverages — even when no reflux event has occurred. The dietary reactivation pathway is as important to manage as the reflux events themselves.
Frequently Asked Questions
What exactly is pepsin and what does it do?
Pepsin is a digestive enzyme produced in the stomach whose job is to break down proteins from food into smaller fragments called polypeptides, which can then be further digested in the small intestine. It starts as an inactive molecule called pepsinogen, is activated by stomach acid, and is most active at the highly acidic pH values (1.5–3.0) found in the stomach. The problem for reflux sufferers arises when pepsin travels outside the stomach via reflux into tissues it is not designed to reach.
Why is pepsin more important than acid in LPR?
Because laryngeal tissue is essentially resistant to damage from acid alone at the pH values that characterise reflux reaching the throat — but is significantly damaged when pepsin is present at the same pH. Direct tissue studies have confirmed this: laryngeal tissue exposed to pH 4 acid without pepsin shows minimal damage; laryngeal tissue exposed to pH 4 with pepsin shows substantial damage. The throat’s vulnerability is to the enzyme, not the acid alone. This explains why acid-suppressing medications (PPIs) consistently fail to resolve LPR throat symptoms.
Can pepsin be reactivated by food and drink?
Yes — this is one of the most clinically important facts about pepsin in LPR management. After a reflux event deposits pepsin in the throat, that pepsin becomes dormant at the throat’s natural pH of 6.8. But anything that drops the local pH below 6.5 — an acidic drink, citrus food, carbonated beverage, even coffee — reactivates the dormant pepsin, triggering further tissue damage without a new reflux event. This is why dietary acid management in LPR is about more than preventing new reflux.
Do PPIs get rid of pepsin?
No. PPIs reduce acid production by blocking proton pumps in gastric parietal cells. This reduces pepsinogen activation (because less acid means less conversion from pepsinogen to pepsin), but it does not inactivate or remove pepsin that has already been activated. Refluxate in PPI-treated patients typically has a higher pH but still contains active pepsin — and pepsin at mildly acidic pH (pH 4, characteristic of PPI-treated patients) still causes significant tissue damage through E-cadherin cleavage and barrier disruption.
How does pepsin get inside cells?
Pepsin is taken up into laryngeal epithelial cells by a process called receptor-mediated endocytosis — it binds to specific receptors on the cell surface and is internalised in membrane-bound vesicles called endosomes. Endosomes are naturally acidic (around pH 5), which reactivates the pepsin once it’s inside the cell. From there, it can damage intracellular structures, deplete protective stress proteins, and trigger inflammatory signalling cascades — all from inside the cells of the laryngeal lining.
What permanently destroys pepsin?
Pepsin is permanently and irreversibly denatured (destroyed) at pH ≥8. The 2012 Koufman and Johnston study demonstrated that naturally alkaline water at pH 8.8 instantly and irreversibly inactivated human pepsin. Below this threshold — including at the neutral pH 6.7–7.4 of ordinary drinking water — pepsin is not destroyed but preserved in a dormant state. This is why alkaline water above pH 8.8 is specifically relevant to pepsin management in LPR, and why ordinary water has no meaningful effect on pepsin stability.
Can pepsin be detected to confirm I have LPR?
Yes — salivary pepsin testing using the Peptest device can detect pepsin in saliva samples. Because pepsin can only reach the saliva via reflux from the stomach, a positive test confirms that reflux has reached the throat or upper oesophagus. The test has high specificity (around 98% in fasting samples for LPR) but lower sensitivity, making it most useful as a confirmatory tool rather than a standalone diagnostic. It is also non-invasive and can be done at home, which gives it practical advantages over pH impedance monitoring for initial investigation.
Conclusion
Pepsin is the missing piece in most people’s understanding of acid reflux — and particularly in understanding why LPR is so much harder to treat than standard GERD. It is not just an incidental passenger in reflux; it is the primary damaging agent for throat and laryngeal tissue, operating through mechanisms that include direct proteolytic attack on mucosal proteins, intracellular infiltration via receptor-mediated endocytosis, depletion of the larynx’s own protective proteins, and a reactivation cycle that extends the inflammatory damage far beyond each individual reflux event.
Understanding pepsin explains why PPIs so consistently disappoint LPR patients — they reduce the acid that activates pepsin but don’t touch the enzyme itself. It explains why dietary acid management is not optional extra advice but a mechanistic necessity. And it explains why the multi-layer approach — reducing reflux events, physically blocking pepsin with alginates, inactivating it with alkaline water, and avoiding dietary reactivation — works in ways that acid suppression alone simply cannot.
If you’re building an evidence-based approach to managing your reflux with this understanding in place, the Wipeout Diet Plan translates the pepsin biology into a practical structured protocol — covering the dietary and lifestyle framework that addresses reflux at the source. The Wipeout Food Reference Guide gives you the pH values and reflux potential of individual foods and drinks — the essential companion for applying the dietary reactivation knowledge in daily practice.
Research Sources
[__Johnston et al., The Laryngoscope, 2007__] — Definitive study establishing the pH-dependent activity and stability profile of human pepsin 3b; confirmed that pepsin is active up to pH 6.5, stable and dormant at pH 7.4 for at least 24 hours, and that 72% of peptic activity is recoverable after reactivation from neutral pH by a drop to pH 3; established the laryngopharyngeal pH of 6.8 as a pepsin-preserving environment and the theoretical basis for dietary acid avoidance in LPR.
[__Johnston et al., Annals of Otology, Rhinology and Laryngology, 2009__] — First study demonstrating that pepsin at pH 7.4 (non-acidic reflux conditions) causes cell damage in hypopharyngeal epithelial cells; confirmed receptor-mediated endocytosis as the uptake mechanism; documented mitochondrial swelling, cytotoxicity, and dysregulated gene expression at neutral pH; proposed pepsin receptor antagonists and irreversible pepsin inhibitors as future therapeutic targets.
[__Bulmer et al., The Laryngoscope, 2010__] — Experimental study using excised porcine laryngeal tissue in Ussing chambers demonstrating that laryngeal tissue is essentially resistant to damage at pH 4.0 alone, but is significantly damaged when pepsin is present at the same pH; established that pepsin, not acid, is the primary damaging agent in LPR — the single most important mechanistic finding for understanding why acid suppression alone fails to protect laryngeal tissue.
[__Johnston et al., Annals of Otology, Rhinology and Laryngology, 2006__] — Demonstrated that pepsin causes depletion of laryngeal protective stress proteins Sep70 and Sep53 through receptor-mediated endocytosis; in LPR patients, Sep70 was significantly depleted (p=0.027) compared to controls; confocal microscopy confirmed intracellular pepsin uptake; concluded that this altered stress protein response leads to cellular injury and plays a role in LPR disease development.
[__Gill et al., Annals of Otology, Rhinology and Laryngology, 2006__] — Immunohistochemistry and Western blot analysis of 54 laryngeal biopsy specimens from 18 LPR patients; detected intracellular pepsin in LPR patients but not controls; found reduced E-cadherin expression in LPR patients; established the inverse association between pepsin presence and laryngeal protective mechanisms (CAIII, E-cadherin) as a hallmark of reflux-mediated laryngeal injury.
[__Samuels et al., International Journal of Molecular Sciences, 2023__] — Cell study demonstrating that alginate medications (Gaviscon Advance and Double Action) protect against pepsin-mediated E-cadherin cleavage and MMP induction in Barrett’s oesophageal cells at pH 4; identified regulated intramembrane proteolysis as a novel pepsin injury mechanism; demonstrated lasting protection from alginate even after washout; supported topical protection as a therapeutic approach for GERD and LPR.
[__Koufman & Johnston, Annals of Otology, Rhinology and Laryngology, 2012__] — Established that pH 8.8 naturally alkaline artesian well water instantly and irreversibly inactivates human pepsin and has superior acid-buffering capacity compared to neutral waters; demonstrated the threshold above which pepsin is permanently denatured rather than merely suppressed; formed the scientific basis for alkaline water as an adjunct in reflux management, particularly for LPR.
[__Li et al., Frontiers in Medicine, 2025__] — Comprehensive review of pepsin-induced GERD pathogenesis; documented the cascade of pepsin-mediated injury mechanisms including tight junction degradation (Claudin-1, ZO-1, occludin), E-cadherin cleavage via the ROS/ERK/c-Jun/MMP pathway, and widening of epithelial intercellular spaces; synthesised evidence for pepsin as an independent molecular driver of GERD severity separate from its effects in LPR.
[__StatPearls, Physiology, Pepsin, NCBI Bookshelf__] — Authoritative clinical reference covering pepsinogen synthesis and secretion by gastric chief cells, pepsin activation by HCl, the autocatalytic activation mechanism, and pepsin’s role as an endopeptidase cleaving peptide bonds in dietary proteins; establishes the foundational physiology underlying all clinical applications of pepsin biology to reflux management.
David Gray
Content Researcher & Author
David Gray founded Wipeout Reflux to address a critical gap in reflux management. His research synthesizes over 100 peer-reviewed studies on laryngopharyngeal reflux (LPR), pepsin biology, and GERD pathophysiology. For LPR specifically—a condition most physicians misdiagnose—his work focuses on pepsin reactivation and why standard PPI therapy fails most patients. He develops evidence-based protocols targeting root causes of both LPR and GERD, integrating emerging research on sphincter dysfunction, dietary interventions, and newer clinical approaches. Wipeout Reflux represents practical application of clinical science for patients seeking real solutions.